
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
蒽醌-2-羧酸作为光催化剂催化解聚木质素
[廖声甫1, 2, 3, 4 , 刘琪英1, 2, 3, 4 , 马隆龙1, 2, 3, 4†  ]
]
]
|
|


作者简介:廖声甫(1995-),男,硕士研究生,主要从事木质素光催化解聚研究。马隆龙(1964-),男,博士,研究员,博士生导师,主要从事生物质催化利用研究。
木质素是自然界中丰富的可再生芳香碳资源,其解聚得到的单体可以作为重要的化工原料。以蒽醌-2-羧酸作为光催化剂,在硝基苯存在和LED光源照射下,5 h内木质素模型化合物中 β-O-4键有80%的转化率。对于木质素 β-O-4多聚体,该体系也表现出了光催化活性,将蒽醌-2-羧酸负载在非均相载体上,在光催化降解中也可以获得77%的底物转化率。该反应涉及了木质素 β-O-4中C α—C β键和C β—O键的断裂,在催化剂的作用下,首先发生C α—OH的脱氢,随后经过分子内的断键和重新成键生成苯甲醛和愈创木酚。本研究加深了对光催化木质素氧化过程中C—C键断裂过程的认识,有助于理解木质素的解聚机制。
Lignin is an abundant renewable natural resource of aromatic carbon resource in nature, and the monomer obtained by its depolymerization can be used as an important chemical raw material. In this paper, anthraquinone-2- carboxylic acid was used as the photocatalyst for the depolymerization of lignin model compound. In the presence of nitrobenzene and LED light source, 80% conversion of lignin β-O-4 bond was realized within 5 hours. This system also showed photocatalytic activity towards the depolymerization of lignin β-O-4 polymer. By loading anthraquinone-2- carboxylic acid on a heterogeneous carrier, 77% substrate conversion was delivered. In this reaction, cleavage of the C α—C β bond and C β—O bond in the lignin β-O-4 model was involved, the dehydrogenation of C α—OH occurred firstly, and then benzaldehyde and guaiacol were generated by bonds breaking and re-bonding in the molecule. This study gives a deeper understanding of the C—C bond breaking process in the photocatalytic lignin oxidation process, which helps us understand the mechanism of lignin β-O-4 bond depolymerization.

开放科学(资源服务)标识码(OSID)
木质素是一种复杂的植物源性生物聚合物, 约占所有陆地生物质的20%[1, 2]。与纤维素和半纤维素相比, 木质素的高度交联聚合结构使得其解聚尤为复杂, 因此木质素通常被当作废弃物焚烧处理, 只有大约5%的木质素被低级利用[3], 例如作为低级燃料[4]和混凝土添加剂[5]。木质素由三种苯丙烷单体(紫丁香基、愈创木基和对羟苯基)通过C— C键和醚键连接而成[6, 7], 是自然界中唯一的可再生芳香化合物来源[8, 9]。在植物细胞中, 木质素作为细胞壁的主要成分, 保证了植物的刚性。木质素的结构决定了其化学惰性, 可以更好地连接纤维素和半纤维素以及保护植物。
在众多木质素连接键中, 以β -O-4连接键最多, 通常占所有木质素连接键的50%以上[10, 11]。其他几种常见的木质素连接键有4-O-5、β -5、5-5、β -β 、β -1等[12, 13]。因此, 有效地断裂木质素β -O-4连接键是解聚木质素的关键。
光催化解聚木质素首先由STEPHENSON等提出。2013年, STEPHENSON和同事研究了光催化解聚木质素的可能性[14]。他们首先通过光催化还原裂解β -O-4酮的C— O键上, 以 [Ir(ppy)2(dtbbpy)]PF6作为有效的光催化剂, 在12 h内得到了4′ -甲氧基苯乙酮和愈创木酚, 收率分别为88%和89%。在这个过程中愈创木酚没有发生任何氧化, 且产物之间没有发生聚合。他们的后续研究通过结合可见光光氧化还原催化和Pd催化建立了用于氧化反应步骤的光催化方案[15]。过硫酸钠(Na2S2O8)被用作廉价的终端氧化剂, 并与Ir络合物引发的光催化循环结合。该体系与模型化合物和富电子芳烃均能很好地配合, 得到的木质素Cα — OH氧化产品收率为83% ~ 96%。
两步法氧化还原解聚木质素的一个挑战是氧化反应和还原反应不相容。为此, LUO等[16]采用双波长切换策略, 以Pd/ZnIn2S4和TiO2分别作为Cα — OH氧化为Cα =O和Cβ — O裂解的光催化剂, 采用一锅法, 使用双波长的LED照射, 获得大于90%的单体产率。
上述的逐步氧化-还原解聚分别需要外部氧化剂和还原剂, 从原子经济的角度来看是不利的。因此, 催化转移氢化(catalytic transfer hydrogenation, CTH)可能是消除外部氧化还原剂需求的理想方法。基于此, LUO等[17]报道了一种催化剂和可见光驱动的CTH方案, 这是木质素破碎的整体可持续过程。他们利用ZnIn2S4作为非均相光催化剂来促进自氢转移氢解木质素模型化合物和有机溶剂木质素直接转化为酚醛产物。王野团队[18]也证明CdS具有同样的效果。这为木质素的光催化解聚提供了一种新的思路。
蒽醌(anthraquinone, AQ)是一种重要有机染料前体, 很多染料都由蒽醌衍生而来, 例如茜素[19]。蒽醌被用作染料的历史由来已久, 在许多历史织物和绘画中都检测到其踪迹[20], 这些颜料在色调和透明度方面都比矿物基颜料具有更好的灵活性[21]。另外, 蒽醌还被用作造纸工业中的消化助剂[22], 在碱性制浆过程中, 9, 10-蒽醌通常被作为氧化还原催化剂加入其中, 用来氧化纸浆中的多糖(纤维素和半纤维素)以保护其不被碱性腐蚀。在此过程中蒽醌被还原成9, 10-二羟基蒽并与木质素发生反应, 使得木质素在水中的溶解度升高, 更容易被洗脱。
蒽醌的一个大规模的工业应用是过氧化氢(H2O2)生产。当前H2O2的主要商业生产方法是蒽醌法[23]。在蒽醌工艺中, 蒽醌通过加氢工艺和自氧化工艺两个过程催化空气中的O2将H2氧化为H2O2。
除了热催化H2O2生产, 近年来, 蒽醌作为光敏剂[24]或者单独的光催化剂[25]在光催化领域也崭露头角。XU等[26]研究发现, 2-乙基蒽醌同样可以作为光催化剂, 应用于连续的光催化H2O2生产。该过程以有机溶剂作为氢供体, 避免了高纯度氢气的使用, 且H2O2生产中的氢化和自氧化采用一锅法进行, 减少了工艺步骤, 具有很高的工业参考价值。另外, 蒽醌在各种有机氧化还原反应中也表现出优异的光催化性能, 例如C— H键的碘化[27]、溴化[25]、氟化[28]以及氧化[29]。除了作为光氧化催化剂外, 蒽醌还可以作为光还原催化剂, BARDAGI课题组报道了1, 8-二羟基蒽醌用于芳基卤化物还原活化的研究[30], 通过蒽醌的还原作用, 将芳基卤化物还原成自由基, 然后用捕获剂进行官能化。这些进展都说明蒽醌类光催化剂具有很好的电子转移能力, 是优异的氧化还原反应催化剂。
本研究以木质素β -O-4模型物作为研究对象, 分析木质素β -O-4键在光催化过程中的变化。β -O-4键是木质素中含量最多的单体连接键, 因此β -O-4模型物相较于其他木质素模型化合物更能代表真实木质素中单体的连接情况。但真实木质素是个十分复杂的多聚体, β -O-4模型物并不能完全模拟真实木质素在解聚过程中的变化。采用蒽醌衍生物作为均相光催化剂, 在硝基苯作为氧化剂条件下探究木质素β -O-4键的断裂情况。在这个过程中会发生C— C键和C— O键的断裂。为解释木质素氧化解聚过程中这些现象, 本文进行了一系列探究实验, 以期归纳出光催化木质素解聚的自由基反应机理。
1.1.1 2-(2-甲氧基苯氧基)-1-苯基乙醇
2-(2-甲氧基苯氧基)-1-苯基乙醇采用文献报道方法合成[17, 31]。取25 mmol 2-溴苯乙酮和45 mmol无水K2CO3溶于100 mL丙酮中, 随后加入30 mmol愈创木酚, 将混合物在80℃下搅拌回流, 反应3 h后, 取出得到的混合物, 过滤后保留液相部分, 蒸去溶剂, 得到淡黄色的β -O-4酮粗产品。将所得的粗产品用乙醇重结晶, 得到白色针状固体(5.2 g, 87%), 将所得固体放入60℃烘箱中干燥8 h后备用。
在β -O-4酮加氢过程中, 取上一步得到的固体6.2 mmol(约1.5 g)加入35 mL四氢呋喃和H2O的混合溶剂中, 四氢呋喃和水的体积比为4∶ 1, 搅拌至混合均匀。随后缓慢加入0.94 g NaBH4, 在室温下搅拌反应3 h后, 用饱和NH4Cl溶液淬灭反应。将混合溶液用乙醚萃取三次, 合并有机相, 将有机相用旋转蒸发仪旋干溶剂, 得到黏稠状的液体(1.24 g, 84%), 放入真空干燥箱中60℃干燥数小时, 密封移入冰箱的冷藏室保存。
1.1.2 木质素多聚体
木质素多聚体的合成参照文献[8]的方法进行。在装有磁力搅拌子的50 mL圆底烧瓶中加入起始单体2-溴-1-(4-羟苯基)乙酮(1.68 g, 7.86 mmol)和K2CO3(1.63 g, 11.8 mmol)。在氩气气氛下用注射器将10 mL无水二甲基甲酰胺(dimethylformamide, DMF)加入烧瓶中。50℃下加热4 h。反应开始时, K2CO3是反应混合物中唯一的非均相物质。约10 min后, 反应变得浑浊, 1 h内反应变得非常剧烈, 表明形成了聚合物, 该聚合物迅速析出, 之后再添加5 mL DMF以促进充分搅拌。反应停止, 将反应混合物冷却至室温, 然后将混合物倒入加冰的去离子水(100 mL)中, 并使用1 mol/L HCl酸化至pH值为2 ~ 3。酸化导致混合物从橙色变成浅橙色-黄色。然后将该混合物过滤, 用去离子水和乙醇洗涤数次, 收集聚合物并通过冷冻干燥机干燥8 h, 得到橙黄色粉末状固体(0.906 g, 85%)。
在装有磁力搅拌子的100 mL圆底烧瓶中加入上一步得到的聚合物(0.476 g, 3.5 mmol)和25 mL无水二甲基亚砜(dimethyl sulfoxide, DMSO), 所得混合物放入油浴锅中搅拌。将NaBH4(0.59 g, 14 mmol)分批加入聚合物混合物中, 通常会观察到起泡和轻微的颜色变化。加入NaBH4后, 将圆底烧瓶用磨口玻璃塞密封并加热至50℃, 搅拌反应24 h。随着反应的进行, 混合物的溶解性增大。反应结束后, 将反应混合物转移至200 mL加冰的去离子水中, 搅拌下加入2 mol/L HCl, 将混合物酸化至pH值为2 ~ 3, 然后将反应混合物过滤, 并用去离子水和乙醇洗涤数次。收集所得固体, 并在真空干燥箱中干燥5 ~ 12 h, 所得产物即为β -O-4聚合物模型物(0.386 g, 81%)。
1.2.1 C3N4催化剂
C3N4催化剂根据文献[31]方法合成, 通过在550℃下加热三聚氰胺得到C3N4催化剂。具体操作如下:称取10 g三聚氰胺放入带盖的陶瓷坩埚内, 并将其移入空气马弗炉内, 以2.5℃/mim的升温速率从室温升至550℃, 然后保持3 h。待冷却到室温后, 将坩埚从马弗炉中取出, 得到的黄色固体依次用去离子水和乙醇清洗数次, 以除去未反应的三聚氰胺。将得到的黄色粉末放入70℃烘箱中干燥8 h, 从而制得C3N4催化剂。
1.2.2 C3N4负载的蒽醌复合催化剂
AQ-C3N4通过胺与羧酸的直接耦合合成[32]。具体步骤如下:在圆底烧瓶中加入200 mg C3N4、20 mg蒽醌-2-羧酸、5 mg CrCl4和20 mL甲苯, 混合物在50℃下搅拌反应12 h。反应结束后, 将混合物冷却至室温, 过滤, 将所得固体用去离子水和乙醇洗涤数次, 在70℃烘箱中干燥8 h, 得到AQ-C3N4复合型催化剂。
1.2.3 壳聚糖负载蒽醌催化剂
AQ-壳聚糖(AQ-Chitosan)催化剂采用与AQ-C3N4类似的方法合成。将200 mg商购壳聚糖和20 mg蒽醌-2-羧酸和5 mg无水Na2SO4置于圆底烧瓶中, 并加入10 mL乙腈, 在50℃下搅拌反应12 h, 过滤, 将所得固体用去离子水洗涤数次, 在70℃下干燥8 h, 即得到所需的AQ-Chitosan催化剂。
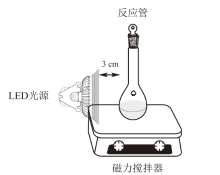
木质素解聚在实验室自制的光催化反应器中进行, 如图1。标准的蒽醌光催化木质素β -O-4模型物解聚过程具体操作如下:取木质素模型化合物(0.1 mmol)、蒽醌类催化剂(0.01 mmol)、硝基苯(0.1 mmol)放入玻璃反应管中, 加入3 mL乙腈作为溶剂, 密封, 通过5次抽气-充气循环, 用氩气替换管内的空气。然后在搅拌下用30 W紫色LED光源(波长为400 nm左右)照射反应装置, 开始计时。到达设定的反应时间后, 关闭光源, 用溶剂稀释3倍的同时, 加入内标物乙酰丙酸乙酯, 留待分析。
 | 图1 自制的光催化反应器Fig. 1 Homemade photocatalytic reactor |
木质素解聚的相关实验产物检测和结果分析在配备有HP-5毛细管色谱柱(25 m × 0.32 mm, 0.17 μ m膜厚, Hewlett Packard, CA)和火焰离子化检测器的气相色谱(GC-2014, Shimadzu, Kyoto, Japan)上进行。使用乙酰丙酸乙酯作为内标物, 乙腈为洗针液。样品在加入气相色谱检测样品瓶前, 先用0.22 μ m的微孔滤膜进行过滤。非极性柱气相色谱的检测条件如下:注射器温度为260℃; 检测器温度为280℃; 升温程序为80℃下保持2 min, 然后以10℃/min的升温速率升至280℃, 在280℃下保持2 min。相关实验的定性分析也在配有HP-5毛细管色谱柱的气相色谱质谱联用仪上进行, 检测条件和气相色谱的相同。所有的实验在相同条件下测试三次, 取平均结果, 每次测试误差在 ± 5%以内。
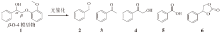
蒽醌具有良好的光催化氧化能力, 因此采用蒽醌类化合物作为光催化剂, 在光照条件下解聚木质素二聚体模型物。使用氧气为氧化剂, 对木质素二聚体进行氧化解聚。图2为木质素β -O-4模型化合物1在氧气环境下的解聚方案, 实验结果列于表1。
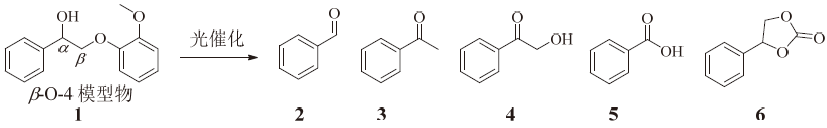 | 图 2 木质素β -O-4模型物氧气解聚方案Fig. 2 Oxygen depolymerization scheme of lignin β -O-4 model compound |
| 表1 氧气环境下的蒽醌光催化解聚木质素模型化合物 Table 1 Anthraquinone photocatalytic depolymerization of lignin model compounds under oxygen atmosphere |
从表1可以看出, 该反应在氧气环境下的转化率很高, 但整体的选择性很低, 且二聚体裂解的酚类部分产物全部降解, 苯基醇类部分收率不到50%。说明蒽醌有催化木质素解聚的潜力, 只是该体系还缺乏一种控制手段, 来调控蒽醌催化剂的选择性。
鉴于氧气环境下解聚反应的低选择性, 有必要对氧化剂进行筛选, 以寻找合适的氧化剂来代替氧气, 这样的氧化剂需要有合适的氧化还原能力。表2列出了不同氧化剂存在下的木质素解聚反应。
| 表2 在不同氧化剂条件下的木质素模型化合物解聚 Table 2 Lignin model compounds depolymerization in the presence of different oxidants |
由表2可知, 当氧化剂为过硫酸铵 [(NH4)2S2O8] 时, 反应主要为化学驱动, 在暗反应或者没有光催化剂的条件下, (NH4)2S2O8也可以氧化木质素模型化合物, 进而发生解聚。木质素在这个过程中主要发生了Cβ — O键断裂, 但是当引入由2-甲基蒽醌触发的光反应后, 不仅能检测到Cβ — O键断裂的产物, Cα — Cβ 键断裂也被检测到了。这说明2-甲基蒽醌的光催化促进了 (NH4)2S2O8的氧化作用, 使得更难断裂的Cα — Cβ 键也发生了断裂, 导致产物分布变多, 选择性低。这说明 (NH4)2S2O8的氧化性比氧气强。接下来尝试使用环保的H2O2作为氧化剂。在经过10 h的照射反应后, 底物转化率为84%, 但两种产物的收率很低, 分别为13%和14%, 且没有检测到愈创木酚生成, 说明酚类在这个条件下也被降解了。这是由于H2O2在紫外光的照射下会产生具有强氧化性的羟基自由基, 导致反应的转化率高而选择性低。当使用硝基苯作为氧化剂时, 在氩气环境下可检测到苯甲醛和愈创木酚两种产物。在这个过程中, 木质素既发生了Cα — Cβ 键断裂, 也发生了Cβ — O键断裂, 说明硝基苯是这个体系中适合的温和氧化剂。
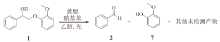
以硝基苯为氧化剂, 对木质素二聚体进行氧化解聚。反应方程如图3所示。硝基苯在反应中充当了氧化剂和质子受体, 在反应过程中逐步被还原。
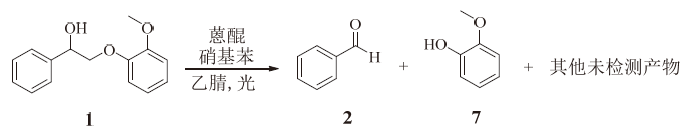 | 图 3 木质素β -O-4模型物在硝基苯存在下的解聚Fig. 3 Depolymerization of lignin β -O-4 model compound in addition of nitrobenzene |
2.3.1 不同催化剂的性能评价
考察了不同光催化剂对木质素模型化合物光催化解聚的效果。结果列于表3。
| 表3 不同催化剂条件下的木质素模型化合物光催化解聚 Table 3 Lignin model compounds degeneration by different photocatalysts |
从表3可以看出, 蒽醌-2-羧酸在所有光催化剂中具有最高的选择性。尽管2-甲基蒽醌、蒽醌和2-溴蒽醌催化木质素解聚的转化率都很高, 但是选择性没有蒽醌-2-羧酸高。2-氨基蒽醌和罗丹明6G在此过程中没有表现出光催化效果, 这是由于蒽醌中芳香环上的氢原子被氨基取代后, 会导致分子荧光增强[33, 34], 从而系间窜越降低, 三重态分子减少, 并且2-氨基蒽醌三重态在乙腈中很快和溶剂分子碰撞湮灭[35]。
2.3.2 木质素光催化解聚中的溶剂效应
几种溶剂被用于评价蒽醌光催化木质素解聚的效果来探究反应的最佳溶剂。反应结果列于表4。
| 表 4 木质素模型化合物光催化解聚的溶剂筛选 Table 4 Solvent effect of photocatalytic lignin model compounds degeneration |
由表4的结果可知, 乙腈中木质素模型化合物解聚获得的产率最高。对于前四种溶剂, 木质素模型化合物的转化率相差不大, 但产率的差异较大。而对于DMF, 有少部分木质素参与了反应, 但没有检测到明显的解聚产物。值得一提的是, 反应结束后发现了苯胺, 且硝基苯完全消失, 说明在DMF中硝基苯会发生还原, 生成苯胺, 从而阻止了其与木质素的反应。在质子性溶剂乙醇中, 木质素也没有发生反应, 这是由于乙醇中含有活泼氢原子, 很容易被氧化成乙醛, 这就和木质素模型化合物的氧化形成竞争, 显而易见, 乙醇氧化成乙醛的反应更容易发生, 阻碍了木质素的氧化。另外, 乙醇在光催化中也可以用作捕获剂, 会抑制光催化反应。乙腈是常见的极性非质子型溶剂, 由于其反应惰性和良好的相容性, 在光催化中常用作溶剂。本实验也选择乙腈作为溶剂。
2.3.3 木质素光催化解聚产物随时间的变化
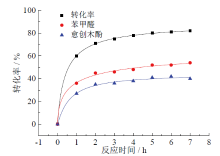
为探究木质素模型化合物在光催化过程的转化率和产率随时间的变化情况, 选取了不同时间点来观察反应, 并将得到的转化率和产率进行非线性拟合, 得到转化率和产率曲线如图4所示。由图可知, 反应在前3 h十分迅速, 之后速率放缓。说明随着底物的消耗, 浓度降低, 反应速率下降。而苯甲醛的产率在任何时刻都大于愈创木酚的产率, 这是由于愈创木酚本身不太稳定, 容易发生分解或聚合。
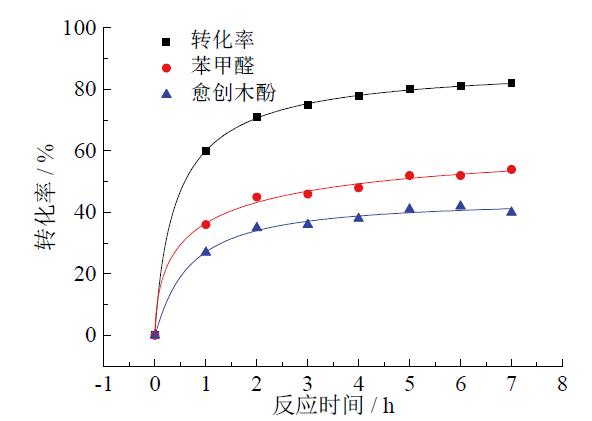 | 图 4 木质素模型化合物解聚产物随时间变化关系Fig. 4 Lignin model compounds depolymerization product over time |
2.3.4 催化反应条件控制
氧化剂的量是影响氧化反应效果的关键因素。本小节探究硝基苯在木质素光催化解聚中的作用, 以考察硝基苯的用量对反应转化率和产率的影响。用不同底物当量的硝基苯进行实验, 考察不加硝基苯、0.33当量、0.67当量、1当量和2当量硝基苯存在下, 木质素转化率和产物分布情况。另外, 光源也是影响光催化反应的重要因素, 寻找合适的光源对反应至关重要, 为此分别选取蓝光和白光这两种可见光源作为对比, 进行光催化测试。反应结果列于表5。
| 表 5 光催化木质素模型化合物解聚控制变量实验 Table 5 Control variable experiments of photocatalytic lignin model compounds depolymerization |
从表5可以看出, 当反应不存在硝基苯时, 蒽醌催化剂也有氧化还原的能力, 但木质素模型化合物的转化率仅为29%, 产率仅分别为6%和10%。加入0.33当量的硝基苯后, 反应的转化率和产率均大幅提高, 说明硝基苯可以有效促进反应的转化和产物的生成。当硝基苯增加到0.67底物当量时, 反应的转化率和产率进一步升高。当硝基苯大于1 当量时, 增加硝基苯的量对反应的转化率和产率的提高很有限, 例如当硝基苯为2倍底物量时, 和1当量的结果相比, 转化率只提高了3%, 产率近乎不变。这说明, 当硝基苯当量为1时, 再增加硝基苯的量得到的效益很小, 经济适用性低, 因此, 1当量是最佳的硝基苯加入量。
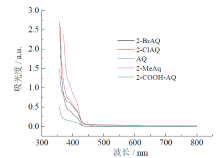
在对反应的光源进行筛选后, 发现紫色LED是效果最好的光源。蓝色LED和白色LED的催化效果均不佳, 即使经过12 h的照射, 转化率也很低或可忽略不计。这是由于特定的光催化剂只能吸收特定波长的光, 仅在特定的波长范围内产生催化效果。从蒽醌类光催化剂的紫外可见吸收光谱(图5)中可以看出, 这些有效的催化剂吸收波长都集中在小于420 nm波长区域, 即紫光和近紫外光, 而对波长大于420 nm的光的吸收几乎为零。这就解释了为什么紫色LED的光催化效果最好, 蓝色LED和白色LED没有效果。
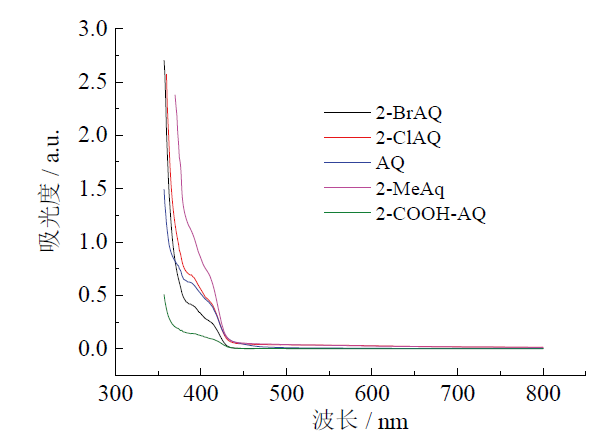 | 图 5 几种蒽醌光催化剂的紫外可见吸收光谱Fig. 5 UV-Vis absorption spectra of various anthraquinone photocatalysts |
2.3.5 木质素β -O-4多聚体模型物的光催化解聚
木质素在植物体内是一种无规则的多聚物。在所有的木质素连接键中, β -O-4连接键占所有木质素连接键的50%以上[10, 11]。为了更好地探究木质素内的键连情况, 合成了由苯丙醇单体共聚形成的β -O-4连接键多聚体。
对于所合成的木质素多聚体, 将其作为底物进行光催化解聚实验, 考察β -O-4键的裂解情况。该实验的方案见图6。
 | 图 6 木质素β -O-4多聚体的光催化解聚Fig. 6 Photocatalytic depolymerization of lignin β -O-4 polymer |
采用称重法称量反应前后的固体重量, 计算得出底物转化率约为50%。气相色谱中检测到了对羟基苯甲醛, 作为该反应唯一的解聚产物, 初步定量为20%左右。这说明木质素多聚体也可以被光催化解聚。在β -O-4键的裂解过程中, 既发生了C— O键的断裂, 也发生了C— C键的断裂。通常, 在木质素β -O-4连接键中, C— O键的键能会低于C— C键的键能, 使其更易于裂解。但是, 在氧化过程中, 键能并不是影响断键的唯一因素, 可能涉及很多方面, 比如分子内电子效应等[36, 37, 38]。
2.3.6 催化剂的固载化及反应效果
均相催化剂最大的缺点是分离困难。为解决这一问题, 可将催化剂进行固载化。
由于C3N4表面的多胺结构, 采用酰胺键的连接方式可将蒽醌-2-羧酸负载在C3N4上。C3N4和负载型催化剂的详细合成步骤见1.2小节。采用相同的方式, 将蒽醌-2-羧酸负载在壳聚糖上, 并将两者进行催化性能比较, 反应结果列于表6。
| 表6 固体催化剂的木质素模型化合物解聚反应效果 Table 6 Result of solid catalyst on the depolymerization of lignin model compounds |
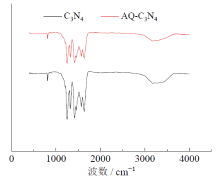
图7为负载前和负载后C3N4催化剂的FTIR图。由图中可以看出, 三嗪单元的变形振动模式归属于798 cm-1处, 而1 250 ~ 1 420 cm-1和1 550 ~ 1 630 cm-1处的吸收峰分别是C— N键和C=N键的伸缩振动[39, 40]。3 050 ~ 3 400 cm-1处的宽吸收峰归属于边缘胺[40, 41]。经过蒽醌-2-羧酸负载后, FTIR图没有发生明显的变化, 但边缘胺的振动强度降低了, 说明经过蒽醌负载后, 边缘胺略有减少。
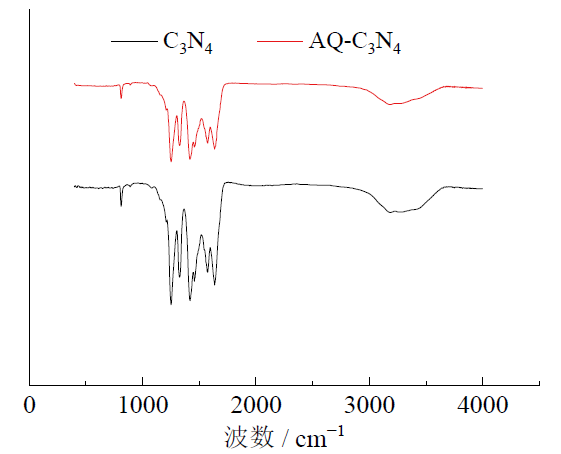 | 图 7 C3N4和AQ-C3N4的傅里叶变换红外光谱Fig. 7 Fourier transform infrared spectra of C3N4and AQ-C3N4 |
从表6中的反应结果可知, 由C3N4固载化的催化剂在经过5 h的照射后可以获得77%的转化率, 并且反应前后催化剂无明显变化。负载在壳聚糖上的催化剂在反应后发生变化而脱落, 由反应前的淡黄色变成初始的白色。说明壳聚糖负载的催化剂不稳定, 无法达到非均相的分离效果。因此, 将蒽醌类催化剂负载在C3N4上, 可以解决均相催化剂分离难的问题。
2.4.1 不同结构木质素底物解聚反应
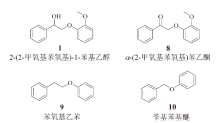
木质素中β -O-4键解聚的一般方法是两步法催化解聚, 最早由STAHL课题组提出[42]。该方法首先对Cα — OH进行氧化, 降低Cβ — O键的键能, 从而使β -O-4键更容易发生解聚。根据相关计算研究, Cα — OH氧化后, Cβ — O键的键能降低了约37.7 kJ/mol, 但是Cα — Cβ 键的键能有所增加, 这导致预氧化后Cα — Cβ 键的断裂更加困难。本研究中, 木质素β -O-4模型化合物不但发生了Cβ — O键的断裂, 还发生了Cα — Cβ 键的断裂。为了探究其断键的机理, 选取了不同形式的木质素模型化合物作为底物, 考察这些化合物在反应前后的变化。具体的模型物和反应结果分别列于图8和表7。
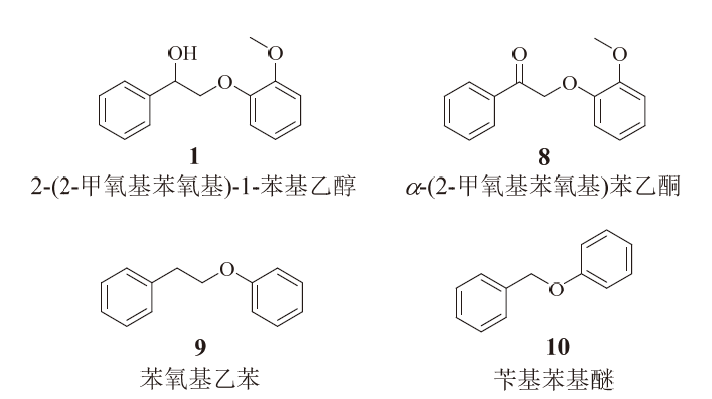 | 图 8 各种木质素模型化合物Fig. 8 Various lignin model compounds |
| 表 7 几种木质素模型化合物的光催化解聚 Table 7 Photocatalytic depolymerization of several lignin model compounds |
对比表7中的反应1和反应2可以看出, 当木质素β -O-4化合物的Cα 上羟基变成羰基后, β -O-4键的断裂效果明显降低, 且没有检测到明显的芳香醛、酚类产物。即使加入乙醇作为外部氢源, 也无法改善反应结果, 如反应3所示, 说明Cα 上的羟基不是充当氢源的作用。另外, 如表7反应5所示, 当Cα 上不存在任何官能团时, β -O-4键的断裂也无法进行, 说明Cα 上的羟基是这个体系中光催化β -O-4键裂解所必需的。在不添加任何光催化剂的条件下, 也有39%的α -(2-甲氧基苯氧基)苯乙酮发生了降解, 说明30%左右的底物降解不是光催化驱动的, 即在这个体系中, 光催化不能有效断裂α -(2-甲氧基苯氧基)苯乙酮中的β -O-4键。表7中的反应6显示, 该体系对木质素的另外一种连接键α -O-4键也有很好的断裂效果。综上所述, 该体系可以直接断裂木质素α -O-4键, 但对木质素β -O-4键的裂解依赖于Cα 上的羟基, 在此体系中, 预氧化对β -O-4键的断裂不能带来帮助。
2.4.2 蒽醌光催化解聚木质素可能的反应机理
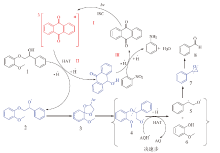
上一小节的实验结果说明, Cα 上的羟基是该体系光催化断裂β -O-4键的必要条件, 可以认定蒽醌光氧化解聚木质素是从Cα 上的羟基开始的。因此以蒽醌为例, 提出了以下木质素β -O-4键的裂解机理, 见图9。
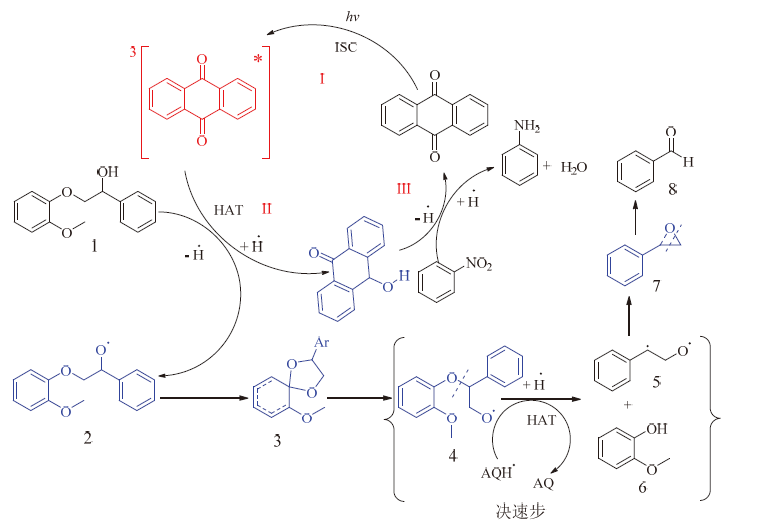 | 图 9 蒽醌光催化裂解β -O-4键可能的机理Fig. 9 Possible mechanism of anthraquinone photocatalytic cleavage of β -O-4 bond |
如图9所示, 蒽醌首先经历过程I, 被光激发和系间窜越, 变成激发三重态, 然后三重态蒽醌分子经过氢转移过程II和木质素底物发生氢转移, 变成氢化蒽醌分子, 氢化蒽醌分子经过程III被硝基苯氧化, 变成了基态蒽醌分子, 完成了催化剂的一个循环。木质素β -O-4模型物1首先被夺去Cα 羟基上的氢, 形成2自由基中间体, 然后由于氧中心自由基的强电负性, 和苯环上的电子发生耦合, 形成中间体3, 中间体3经过一系列分子内电子重排, 变成中间体4, 中间体4会被氢化蒽醌还原, 使得C— O键氢解, 生成愈创木酚6和双自由基5, 双自由基分子中的O和C重新成键, 生成7中间体, 最后中间体7发生C— C键断裂, 生成最终产物苯甲醛。这个机理可以很好地解释了α -(2-甲氧基苯氧基)苯乙酮不发生反应的原因, 主要是由于氢转移是从羟基氢原子开始的。反应结果生成苯甲醛, 而不是苯乙酮。另外, 如表5反应1所示, 在不添加硝基苯的情况下, 木质素β -O-4键也会发生少量断裂, 同时也有少量产物生成, 这是由于氢化蒽醌变成基态蒽醌的方式不仅包括被硝基苯氧化, 在中间体4变成5和6的过程中也可以将氢化蒽醌变成基态蒽醌, 只是该过程相当缓慢, 使得催化剂循环受阻。而当氧化剂为氧化性比硝基苯强的氧气时, 氢化蒽醌来不及将中间体4裂解成5和6, 就会被氧气所氧化, 这就导致在氧气环境下木质素转化率高, 但芳香醛、酚类产率低。
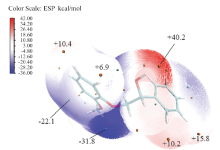
为了进一步验证这个机理, 使用multiwfn[43]和VMD[44]程序对模型物1进行波函数分析, 得出了模型物1的表面静电势分布, 见图10。由图10可知, 模型物1分子表面静电势的极大值点位于Cα — OH处, 这说明羟基上的氢易于解离, 有利于氢转移反应的发生。这印证了该位置是反应起始点的结论。
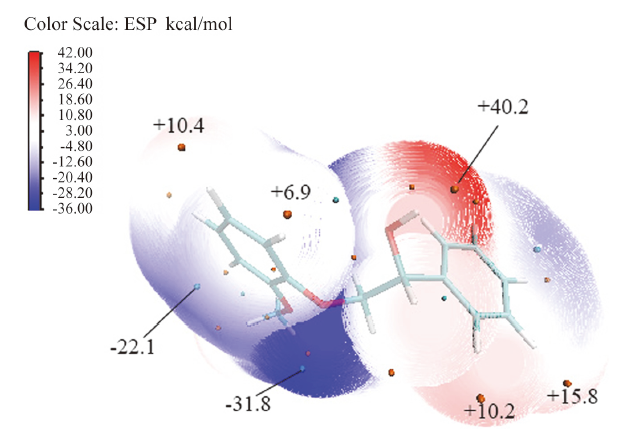 | 图 10 2-(2-甲氧基苯氧基)-1-苯基乙醇的表面静电势Fig. 10 Surface electrostatic potential of 2-(2-methoxy-phenoxy)- 1-phenylethanol |
采用蒽醌衍生物蒽醌-2-羧酸作为催化剂, 在硝基苯的存在下光催化氧化木质素, 5 h内可获得80%的转化率, 获得52%的苯甲醛和41%的愈创木酚单体产率。在光催化过程中木质素β -O-4模型物中发生了C— C键和C— O键的断裂。所提出的自由基反应机理很好地解释了这一现象。但对于由自由基重聚引起的单体产率较低的问题, 尚待下一步研究。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|