

0 引言
“双碳”背景下,清洁能源占一次能源的消费比重日益增加。天然气作为清洁的化石燃料,其重要性日益突出,全球对其需求量持续增长。深海及超深海区域含有丰富的油气资源,是未来能源供应的战略接替区[1]。然而,在深海及超深海区域油气开采时,集输管道面临着极端的高压低温环境,极易出现局部低温和天然气水合物“冰堵”问题,严重影响生产安全与流动保障。
针对水合物堵塞管道问题,常用的防治方法包括伴热、添加化学剂等。其中,水合物动力学抑制剂(kinetic hydrate inhibitors, KHIs)具有用量少、抑制效果好、成本低等优点,受到广泛关注[2-3]。常见的KHIs包括聚N-乙烯吡咯烷酮[poly(N-vinyl pyrrolidone), PVP]、聚N-乙烯基己内酰胺[poly(N-vinyl caprolactam), PVCap]等聚合物[4]。然而,PVP与PVCap等聚合物的生物降解性较差,将增加后期采出水处理的难度与成本。相比之下,氨基酸等天然产物同样具有良好的动力学抑制效果,且易于降解,具有广阔应用前景[5-7]。与利用实验方法选择与评价水合物动力学抑制剂相比[8-13],分子动力学模拟方法能够针对水合物抑制剂实际发挥作用的时间尺度(皮秒至微秒级别)和空间尺度(纳米级别)开展研究,理解其抑制机理及其对水合物形成过程的影响,有助于水合物抑制剂的进一步开发与应用[14-17]。现阶段,通过分子动力学模拟技术研究PVP、PVCap等聚合物以及天然产物类抑制剂对水合物的抑制作用,获得了丰富的信息[14-19]。短肽是由2 ~ 10个具有肽键的氨基酸连接组成,具有相对较小的分子量和简单的结构,其在抑制水合物生成方面具有潜力。目前关于短肽作为水合物抑制剂的研究甚少,且短肽聚合度对水合物生长的影响及其作用机理仍缺乏深入研究。甘氨酸是现有结构最为简单的氨基酸,且与其他氨基酸相比,其对水合物生长具有相对优异的抑制性能[7]。本文利用分子动力学模拟方法,探究甘氨酸及其三肽、五肽对甲烷水合物生长的抑制效果与作用机理,可为开发绿色动力学抑制剂提供理论依据。
1 模型与方法
研究的动力学抑制剂包括甘氨酸单体(glycine, gly)、甘氨酸三肽(3G)、甘氨酸五肽(5G),使用N-乙烯基内酰胺(N-vinyl caprolactam, NVCap)三聚体模拟PVCap,相关物质化学式及分子结构等信息见表1。
表1 PVCap、甘氨酸及其短肽的分子结构Table 1 The molecular structures of PVCap, glycine, and its short peptides |
| 动力学 抑制剂 | 分子式 | 分子结构 | 相对分子质量 |
|---|---|---|---|
| PVCap | C24H41O3N3 | 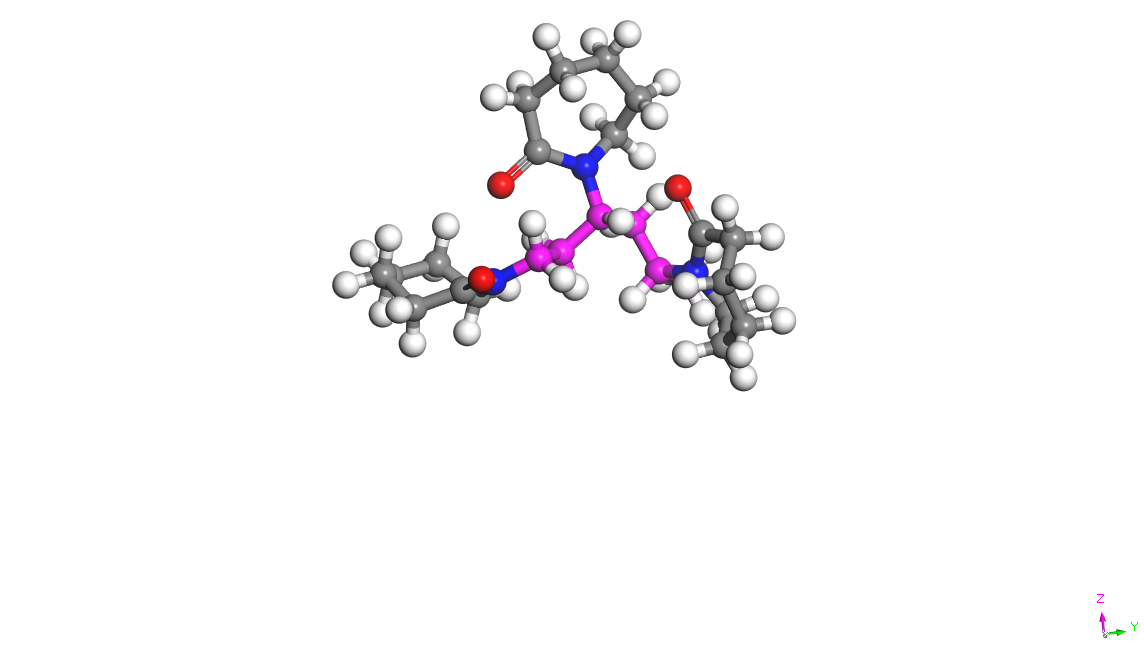 | 419 |
| gly | C2H5O2N | 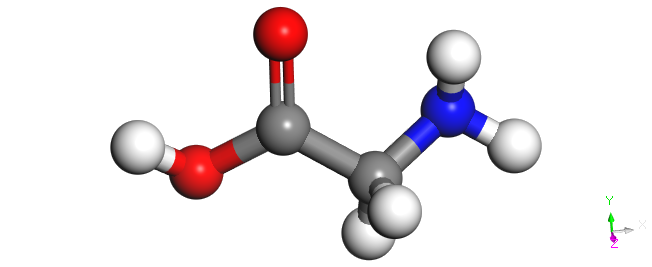 | 75 |
| 3G | C6H11O4N3 | 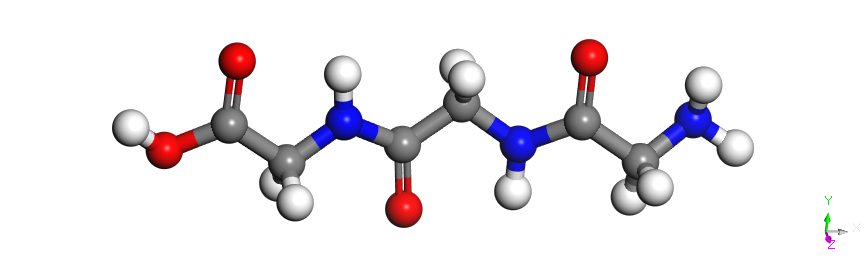 | 189 |
| 5G | C10H17O6N5 | 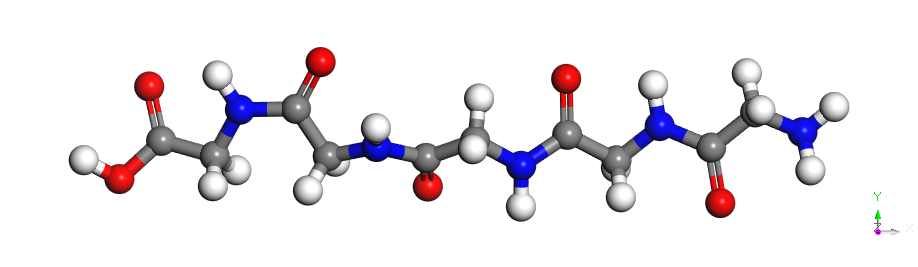 | 303 |
$V(r)=4 \varepsilon\left[(\sigma / r)^{12}-(\sigma / r)^{6}\right]$
式中:V为势能;r为两粒子间的距离;ε为势阱深度;σ为零势能点对应距离。短程相互作用的能量截断半径为1 nm。长程静电力采用粒子−粒子−粒子−网格(particle-particle particle-mesh, PPPM)算法计算[27]。在x、y、z方向规定了周期性边界条件。
2 结果与讨论
2.1 水合物生长构象
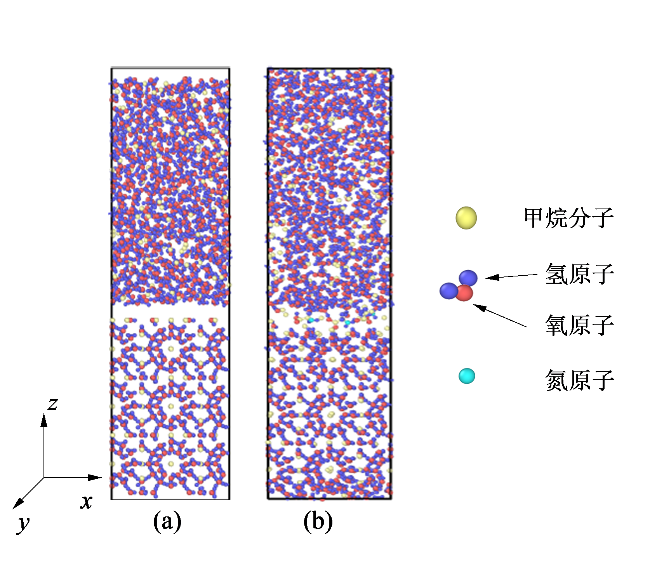
图2展现了不同体系在250 K、50 MPa下甲烷水合物在经过60 ns的动力学模拟后的生长快照。
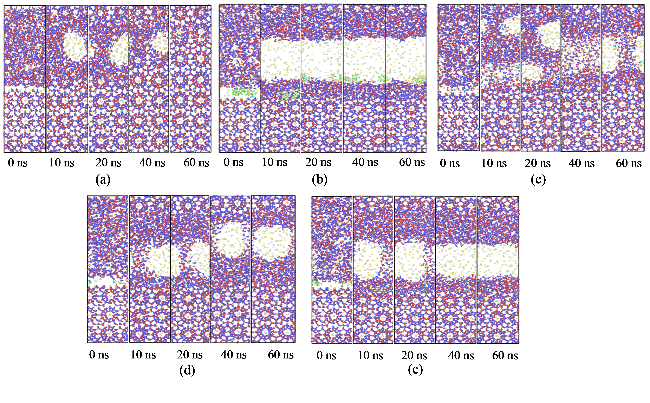
由图2可见,水合物总是沿着水合物晶体表面生长。对空白体系而言,60 ns时水合物接近完全生长状态,甲烷分子与水分子形成的笼型结构基本占据整个模拟箱。由于空白体系中甲烷溶液未受抑制剂的扰动作用,甲烷在水中的溶解度较高,少量甲烷分子聚集形成纳米气泡(10 ~ 40 ns)[图2(a)],分子运动的传质阻力小,扩散速度快。对含抑制剂体系而言,模拟结束时,仍有大量游离的水分子和甲烷,只有少量水合物生成。与空白体系相比,含抑制剂体系中甲烷分子更倾向于聚集而非分散在水分子中,各体系气泡大小依次为PVCap > 5G > 3G > 三个氨基酸单体。特别地,PVCap(10 ~ 60 ns)[图2(b)]与五肽(40 ~ 60 ns)[图2(e)]体系形成了连续气相。抑制剂分子降低了水溶液中甲烷的溶解度,减小形成水合物的驱动力;另一方面,抑制剂分子通过吸附在界面处产生空间位阻,阻碍甲烷分子和水分子运移至水合物界面,有效抑制或延缓了水合物的生长。此外,未在抑制剂附近观察到甲烷和水分子形成笼状结构,说明抑制剂分子对甲烷和水分子的成笼过程具有干扰作用,降低其附近的水分子和客体分子形成水合物的速度。由水合物生长构象可知,甘氨酸五肽与PVCap的抑制性能相近,甘氨酸三肽次之,甘氨酸单体的抑制性能稍弱。
2.2 水合物生长结构参数
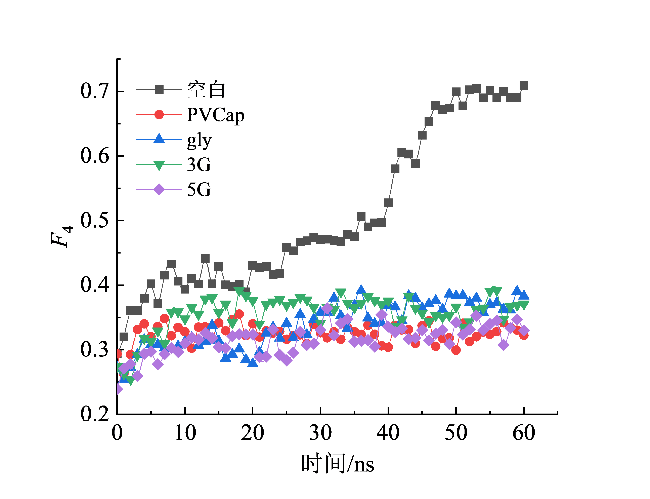
在水合物的动力学模拟过程中,四体结构序参数(four-body order parameter, F4)可用于识别水合物的结晶。F4的计算式如式(2)所示[16]:
式中:φi为两相邻水分子最外侧H原子和水分子中O原子组成的二面角;n为二面角的序号。F4数值随时间上升,说明水合物的生长和结构趋于更加有序,当水合物完全生成后,曲线趋于平衡,F4数值接近0.7,体系中几乎没有自由水分子存在。图4展示了不含抑制剂的空白体系及含抑制剂体系的F4随时间的变化。由图可见,空白体系F4值收敛于0.7,表明随着模拟时间的增加,空白体系中自由水分子数量大量减少,水合物大量生成,且水合物结构变得更加有序。加入抑制剂体系的F4值明显小于空白体系,表明抑制剂的加入明显减缓了水合物的生长速度。在20 ns左右时,含抑制剂体系的F4值基本稳定,没有波动。模拟结束时,含5G体系与含PVCap体系的F4数值基本接近,说明两者对甲烷水合物生长的抑制效果接近。
水合物笼数目能够反映水合物的生成情况。为进一步探究抑制剂对水合物生长的影响,统计了空白体系及含抑制剂体系在60 ns内水合物笼数目随时间的变化以及各体系中不同类型笼数目变化,如图5所示。从图5(a)中发现,与抑制剂体系相比,空白体系水合物笼数目增长速度较快,且数量较多,表明水合物大量生长。图5(b)显示了空白体系各类笼数目随时间的变化,发现还生成51263、51264、51265这三种笼型。51265是不规则笼型,对于不规则笼型出现在SI型甲烷水合物生长过程中的现象在其他文献中也有类似描述[29-30]。51263可以同时存在于SI和SII型水合物中,51264是SII水合物中的笼型。51264笼型出现在甲烷水合物的生长过程中,说明在SI型甲烷水合物的生长过程中也伴随着少量SII型水合物的形成。其他学者在SI型甲烷水合物生长的研究中也观察到类似的现象[26,31-32]。图5(c ~ f)则显示加入抑制剂后,各类笼子数目增长速率有所减慢,并且含抑制剂体系中不再有51264笼型结构生成,说明抑制剂对SI型(51262)与SII型(51264)水合物笼的生长均具有抑制作用。
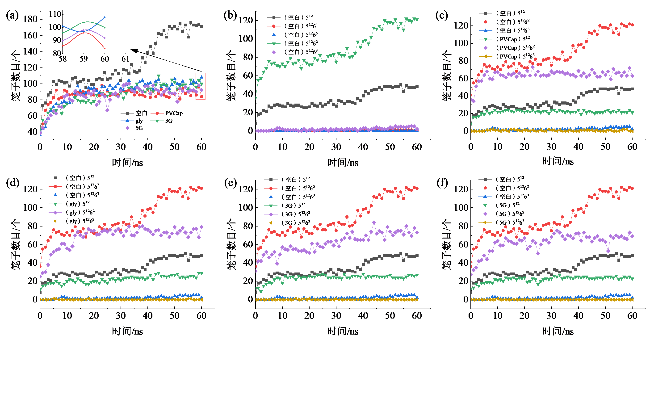
图5 水合物笼子数目随时间变化:(a)不同体系水合物笼子总数;(b)空白体系不同类型笼子;(c)空白与PVCap体系;(d)空白与gly体系;(e)空白与3G体系;(f)空白与5G体系Fig. 5 Number of hydrate cages over time: (a) total number of hydrate cages in different systems; (b) different types of cages in blank system; (c) blank and PVCap systems; (d) blank and gly systems; (e) blank and 3G systems; (f) blank and 5G systems |
2.3 抑制剂作用机理
均方位移(mean square displacement, MSD)、径向分布函数(radial distribution function, RDF)以及氢键分析均可用来表征和理解水合物形成及其动力学和结构特征,能够解释水合物形成和生长过程中的微观物理变化。均方位移曲线体现了体系内粒子偏离最初位置的程度,其曲线斜率通常用于评估分子的扩散系数。其计算式如式(3)所示[33]:
$D_{\mathrm{MS}}=\frac{1}{N} \sum_{i=1}^{N}\left|R_{i}(t)-R_{i}(0)\right|^{2}$
式中:DMS为均方位移参数值;Ri(t) 和Ri(0) 分别为粒子i在t时刻以及初始的位置;N为总粒子数。
当体系中含有大量自由液体或气体时,分子运动受到的限制较小,均方位移参数值随时间的增加而增加;当体系中含有的自由液体或气体较少时,分子运动受到限制,分子在平衡位置振动范围较小。图6显示了不同体系中水分子的均方位移。模拟结束时,空白体系水分子的均方位移曲线斜率变化较小,水分子扩散系数较小,水分子运动受到限制。这表明体系中含有少量自由水分子,水分子运动较慢。而各抑制剂体系中水分子的均方位移曲线斜率较大,水分子扩散系数较大,水分子运动较为剧烈。这表明体系中有大量自由水分子,体系中只有少量水合物生成。进一步说明抑制剂能够有效抑制水合物的生长,其中含PVCap抑制剂体系的均方位移曲线斜率最大,所含自由水分子量最多,抑制性能最强。
径向分布函数表示指定粒子α的位置为参考中心时,粒子β在距离粒子α的r处出现的概率分布大小,通常用于评估系统结构的有序程度。图7显示了不同体系水分子中氧原子之间的径向分布函数,r = 2.75 Å附近存在第一个明显特征峰,代表体系中两个相邻氧原子之间的最近距离是2.75 Å。从图中发现,与空白体系相比,加入抑制剂后峰值降低,体系的有序程度降低,表明体系中含有较多自由水分子,抑制剂的加入有效减缓了水合物的生长。
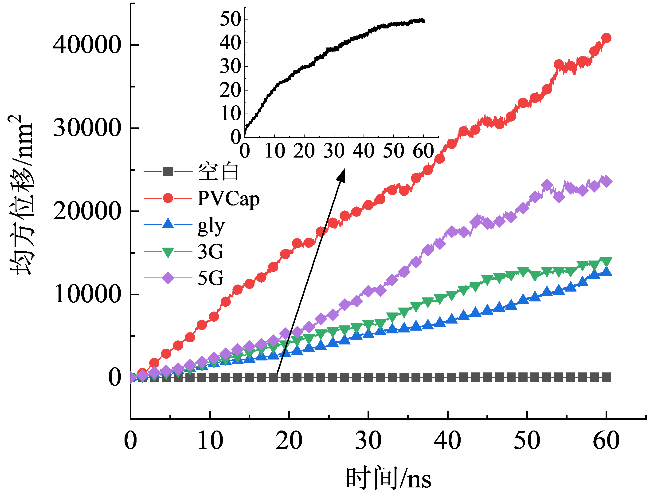
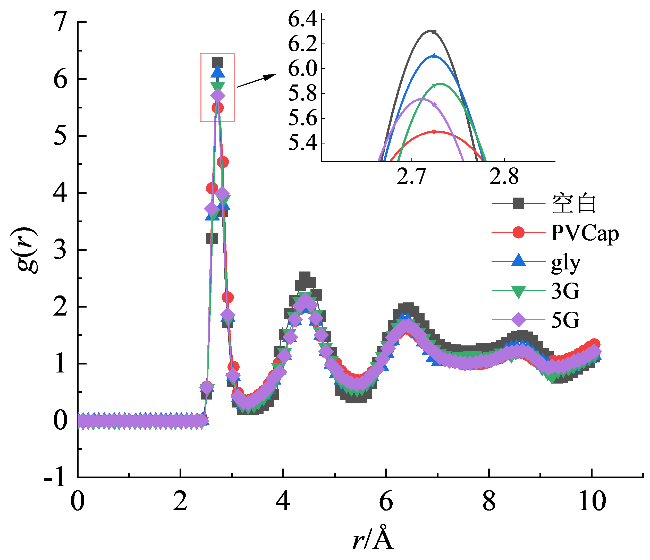
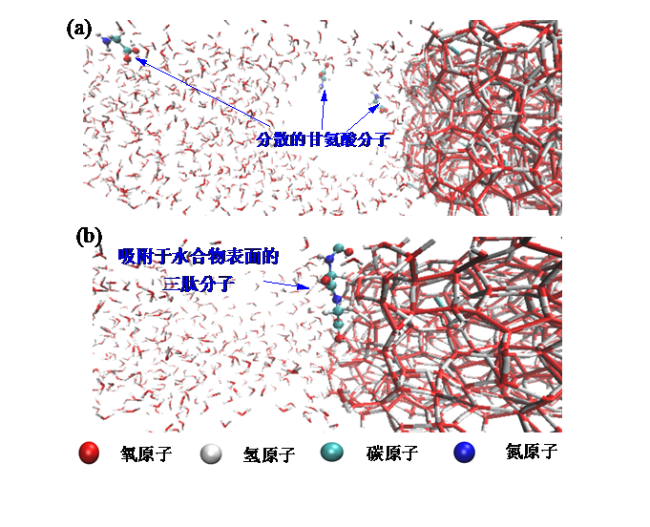
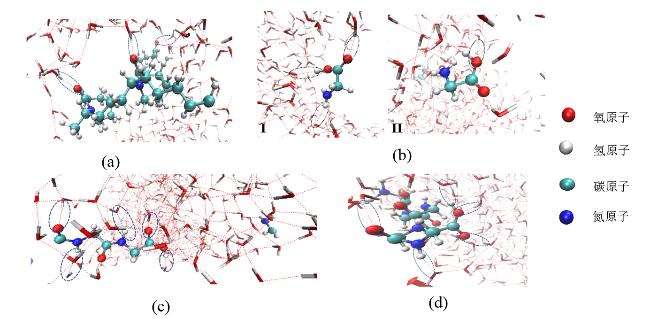
图8为各抑制剂分子与水分子间形成的氢键分析。在图8(a)中,PVCap酰胺基团的双键O原子与水分子的H原子形成氢键,破坏水分子的结构,抑制水合物的生成,这与LIU等[14]的研究结论一致。另外,结合图8(a)和图2(b)发现,在模拟初期,PVCap的七元环倾向于吸附在水合物晶体界面上,增加了空间位阻,阻碍水分子和甲烷分子运移到水合物晶体界面形成水合物,这又与XU等[34]的研究结论一致。这两种作用一方面阻止水分子与甲烷分子运移到水合物界面,另一方面破坏水分子的结构,降低形成水合物的概率。在gly抑制水合物生长的模拟过程中,发现gly与水分子之间有 (I)、(II) 两种成键形式,如图8(b)所示,在 (I)中gly羧基的双键O原子与水分子的H原子形成氢键,羧基的H原子与水分子的氧原子形成氢键;在(II) 中,除了 (I) 中的两种氢键形式,其羧基的单键O原子也会与水分子的H原子形成氢键。这两种成键形式与HU等[18]的研究结论一致。图8(c)显示了甘氨酸的三肽对水合物的抑制作用,三肽中双键O与水分子的H原子形成氢键,与N相连的H原子与水分子的O形成氢键,羧基中H原子与水分子形成氢键,O与水分子的H原子形成氢键。在图8(d)中,甘氨酸五肽与三肽具有相同的成键形式,但五肽具有相对较长的碳链,比三肽、甘氨酸单体能更有效地阻止甲烷和水分子运移到水合物界面上。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 结论
通过分子动力学模拟研究了甘氨酸及其短肽对甲烷水合物生长的影响,并与PVCap进行了比较,得到以下结论:
(1)通过对比构象、径向分布函数等参数,发现甘氨酸五肽的抑制作用与PVCap相近;甘氨酸及其短肽的抑制性能随肽键数增加而增强。
(2)甘氨酸及其短肽通过与水分子形成氢键来抑制或延缓水合物生长;并且甘氨酸与短肽具有相似的成键形式。
(3)甘氨酸与水分子间存在两种形成氢键的形式:羧基上的H原子与双键O原子分别与水分子的O和H原子形成氢键;羧基上的单键O原子与水分子的H原子形成氢键。
(4)PVCap通过七元环吸附在水合物晶体表面,其酰胺基团与水分子形成氢键,抑制或延缓水合物的生长。